Step description¶
The pipeline steps are sumarised in the following figure
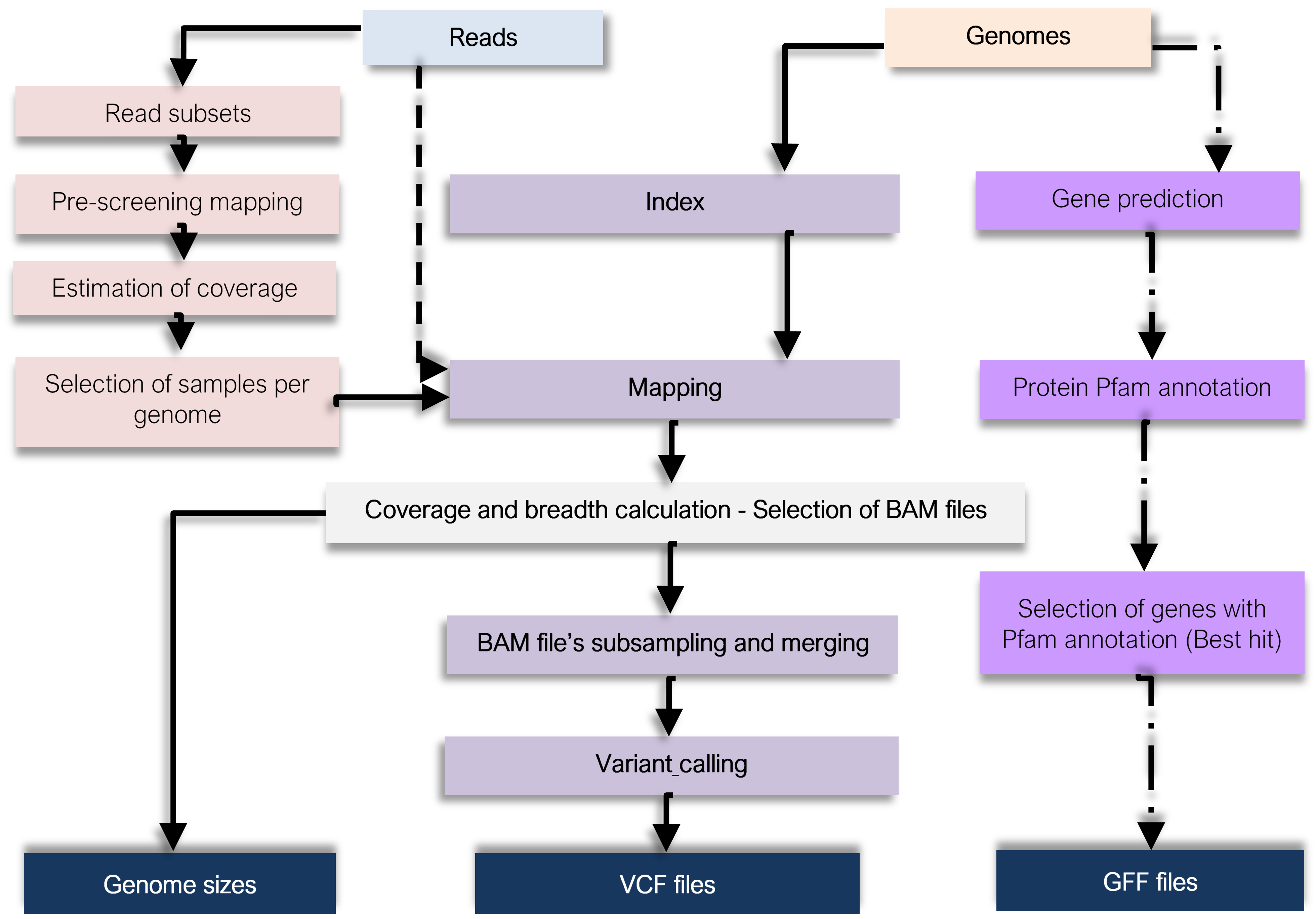
Sample pre-screening¶
This step is realised when the user defines mode: “prefilt” in the pipeline configuration file Input_POGENOM_config.json, as described in section Usage.
A user-defined fraction of paired reads per sample is created using seqtk v1.3. These reads are then mapped to a reference genome, and coverage is estimated: calculated Median coverage/fraction. Samples with Estimated median coverage higher than the user-defined threshold will be kept for further analysis.
Sequence analysis¶
The following paragraph describe the sequence analysis when using default parameters (the user can modify these parameters, as described in section Usage):
Bowtie2 v 2.3.4.3 is used for mapping of metagenome reads to the Metagenome-Assembled Genomes (MAGs). As default, it is required for 95% of the read length to be aligned. The resulting SAM files are sorted and converted to BAM with samtools v1.9. Picards v2.21.6 is used to include sample names in BAM files. Median coverage is calculated using Samtools, ignoring positions that had not acquired any reads. To avoid mapping artefacts such as high coverage of only limited genomic regions, as default ≥40% breadth (fraction of genome covered by at least one read) is needed. It is also wanted a median coverage depth of ≥ 20X (default) to include a sample.
Samples displaying coverage depth values higher than the threshold are downsampled to 20x using Samtools. Freebayes v1.3.1, a haplotype-based variant detector, is used for single nucleotide polymorphism calling. SNP-calling is performed once per MAG, after combining BAM files from the approved samples into a multi-sample BAM file, using Freebayes with the –pooled-continuous flag. SNPs are called when supported by ≥4 reads (default) and with an allele frequency of ≥1% (default). Calls are filtered, removing any sites with an estimated probability of not being polymorphic less than Phred 20 (default, corresponding to a 99% probability of being a real SNP), using vcflib v1.0.0_rc3
Generation of GFF files¶
This step is realised when the user defines annotation: “yes” in the pipeline configuration file Input_POGENOM_config.json, as described in section Usage.
Genes are predicted from contig’s MAGs with Prodigal (v.2.6.3), running the program on each MAG separately in default single genome mode. Predicted genes are annotated against the databases Pfam version 31.0, using hmmsearch version 3.3, and selecting hits with E-value < 0.001. Genes annotated are selected, keeping the best Pfam hit (the highest score), and a GFF file corresponding to those genes is created. This GFF file will be a POGENOM input file.